


- PRESENTATION
- CLASSIFICATION OF CONGENITAL HEART DISEASE
- EPIDEMIOLOGY AND PATHOGENIA
- ORGANIC AND FUNCTIONAL HEART MURMURS
- CLINICAL REPERCUSSION OF HEART DISEASE
- TREATMENT OF CONGENITAL HEART DISEASE
- ACYANOTIC CONGENITAL HEART DISEASE WITH LEFT-TO-RIGHT SHUNT
- ACYANOTIC HEART DEFECTS WITHOUT SHUNTS
- CYANOSIS DEFINITION AND THE DIFFERENCE BETWEEN HIPOXIA AND CYANOSIS
- Cyanotic Congenital Heart Disease
- TETRALOGY OF FALLOT
- PULMONARY ATRESIA WITH VENTRICULAR SEPTAL DEFECT
- D-TRANSPOSITION OF LARGE ARTERIES
- UNIVENTRICULAR HEART
- PULMONARY HYPERTENSION AND EISENMENGER SYNDROME
- DISEASES OF THE ENDOCARDIUM, MYOCARDIUM AND PERICARDIUM
- KAWASAKI DISEASE
- ECG IN THE CHILD
- Arrhythmias in Pediatrics Thisimageismissing
Advances in both diagnostic and surgical cardiac techniques, as well as in medical management of new-borns with severe pathology, have caused an improvement in the survival rate of new-borns and children with congenital heart disease (CHD). Currently, 85% reach adulthood.
Therapeutic action is currently possiblewith all CHDs, and in isolated cases of primary ventricular failure, heart transplants are a feasible option even in the paediatric age range.
CHD is an alteration in the structure of the heart, occurring during the cardiac development of an embryo, which affects its functioning. In addition, a child may present a series of cardiovascular diseases, some of which may require physiopathological knowledge to recognise.
This basic level section is aimed at medical students, paediatric healthcare professionals tending to patients with CHD, and paediatricians and cardiologists in training who want to understand CHD.
Recommended Reading:
There are a great variety of cardiac anomalies and each has the possibility of combining with another, increasing their number considerably. No two CHDs are identical, but there are models which show general similarities.
Various structural defects can be grouped together.In some, a single heart disease that corresponds to a single embryological failure causes all the malformations (such as the Tetralogyof Fallot), although others are more complex and affect several embryological pathways causing issues in different segments and cardiac cavities.
At present, CHDsare classified according to:
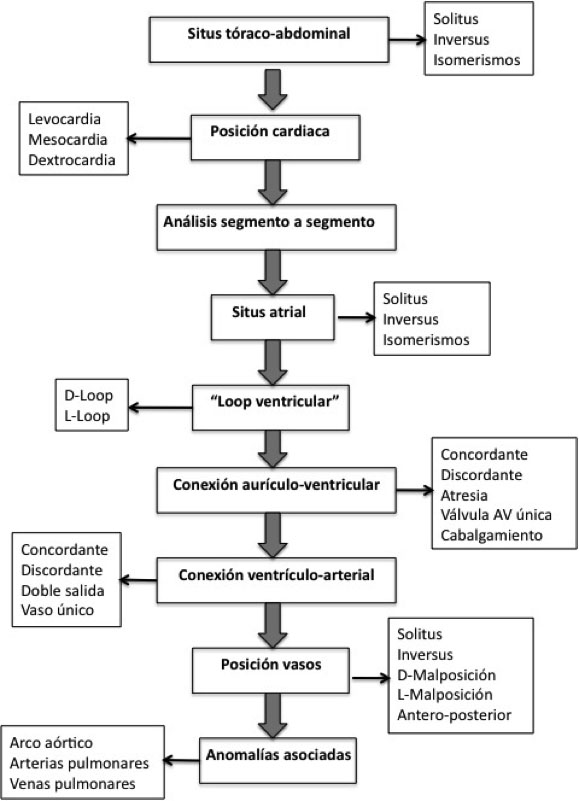
- Anatomy (more typical of cardiologists): : Robert Anderson’s segmental analysis isthe most used nowadays.
CHDs are described by defining the morphology of the atria, ventricles, atrio-ventricular valves and the position of the vessels.

Figure 1: Segmental analysis of the heart
The normal heart is in a position called Situs Solitus, in levocardia and levoapex (where the heart is located in the left hemithorax, with the tip pointing to the left), with atrio-ventricular and ventriculo-arterial concordance (the right atrium connects with the right ventricle which then connects to the pulmonary artery).
The arrangement of the vessels or arteries may be:
- normal (posterior aorta and anterior pulmonary)
- in d-transposition (anterior aorta which is to the right)
- in l-transposition (anterior aorta which is to the left)
- showing vessels in side-by-side position.
There is another classification,described by Dr. Van Praagh (American school), based on the embryological development of the cardiac tube. Cardiac embryology is important in the Van Praagh classification, as is his characteristic 3-letter representation of cardiac anatomy: The normal heart is represented as {S, D, S}
The first letter corresponds to the visceroatrial situs: “S” for situs solitus, “I” forsitus inversus and “A” in isomerisms (heteroataxias, formerly called situs ambiguos).
The second letter shows the direction of the cardiac tube rotation: “D” if it is normal (D-loop, to the right), “L” if it is inverted (L-loop, to the left) and “X” if it is indeterminate.
The third letter pertains to the situation of the vessels: “S” if it is in the normal position with pulmonary artery anterior to the aorta, “I” if the aorta is situated to the left by the pulmonary in the situs inversus,”D” if the aorta is anterior and right, and “L” if it is anterior and left.
- Physiopathology (or functional): CHDs are classified according to their most characteristic clinical presentation. They are divided into acyanotic, cyanotic or mixed forms (they can present cyanosis according to the amount of mixture in the blood and increase in pulmonary flow).

Robert Anderson: Anderson R.H; Ann Thorac Surg 1996: 62: 710-716
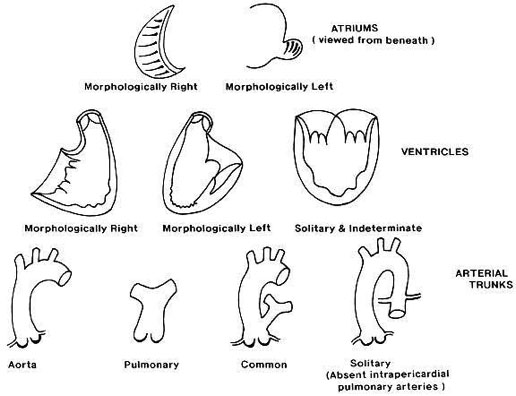
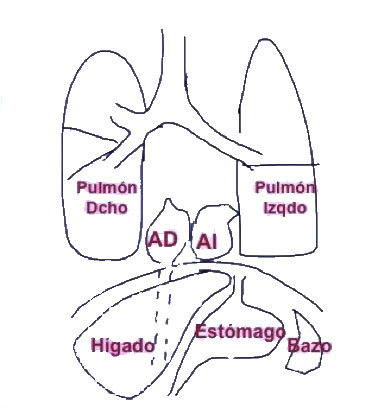
Situs solitus
The visceral situs is the asymmetrical pattern of the body.In the right half of the body we find: the liver and the inferior vena cava (IVC), the appendix, the small intestine and the shorter right bronchus, which bifurcates before the left. On the left side are the spleen, the stomach and the colon. The heart is inside the thorax on the left and with the tip pointed to the left (levocardia and levoapex). The atria are identified by their appendages, the right atrium (RA) is triangularand the left is long, tubular (in glove finger shaped). The situsis identified by its appendages, in general the IVC comes to the RA (note, sometimes there is IVCagenesis) and the pulmonary veins connect to the left atrium. When the liver-IVC-RA triad is on the right, we call thatsitus solitus.
Congenital heart diseases are currently the most frequent cause of death in infants under the age of one. In general, they affect between 5 and 10 per thousand new-borns, of which about a half have clinically significant heart disease and a third may require corrective cardiac surgery at an early age, especially in the neonatal period.
Epidemiological studies suggest that there are genetic factors that contribute to the development of CHD.
In cases with a family history of CHD, recurrence risks are variable depending on: the type of heart disease, whether it corresponds to an isolated malformation or one associated with a syndrome, whether there is an associated maternal disease (like diabetes) or exposure to teratogens (like rubella, alcohol, drugs, etc.)
The human cardiovascular system begins to appear towards the middle of the third week of intrauterine development, when the embryo is no longer able to satisfy its nutritional needs by primitive mechanisms.
Any process that affects the phases of cardiogenesis can cause structural cardiac abnormalities. Many heart diseases have a common embryological cause;therefore, the same heart disease includes several heart defects.
If you want to know more about cardiac embryology, read this article Pediatr Cardiol 2004; 25: 191-200, and look at the following on-line presentations:
There are genetic factors that contribute to the development of heart diseases, including numerical alterations of the chromosome (trisomies, monosomies), structural abnormalities in the chromosome (microdeletions), mutations of one or a pair of genes (autosomal dominant, autosomal recessive or autosomal linked to the X chromosome) and mutations isolated by interacting environmental factors (multifactorial inheritance) [See Table 1]. They can be found accompanying malformations of other organs (more frequent in genetic defects) or isolated (more associated with a multifactorial inheritance pattern).
The study of congenital diseases is currently based on the detection of mutations or polymorphisms in the genes of affected subjects.
Table 1: Percentage Riskof congenital heart disease in the normal population and in relatives with affected cases
| Riego % | |
|---|---|
| General population | 0,5-1% |
| Family 1st degree affected | 2-3% |
| Two siblings or child and parent | 10% |
| More than 2 relatives of 1st degree | Around 50% |
Table 2: The most frequent genetic syndromes associated with heart disease.
SYNDROME – PERCENTAGE -MOST FREQUENTLYASSOCIATED HEART DISEASE
| SYNDROME % | PERCENTAGE | MOST FREQUENTLYASSOCIATED | |
|---|---|---|---|
| CHROMOSOMOPATHIES | Down – (trisomy 21) | 50% | AVD, VSD, ASD, PDA |
| Edward – (trisomy 18) | 100% | VSD,ASD, PDA | |
| Patau – (trisomy 13) | 100% | VSD, ASD, PDA | |
| Turner – (monosomy X) | 25% | AoC, BAo, AoS, HCM | |
| MICRODELECTIONS | Deletion 9p | Frequent | VSD, PDA, PS |
| 4p deletion | Frequent | ASD, PS, VSD | |
| Delection 22q11 (SDM DiGeorge) | 90% | VSD, RAA, FT, PS, CTA | |
| Delection 7q11 (SDM Williams-Beuren) | 100% | Supravalvular AoS, PTS | |
| MUTATIONS | Noonan (PTPN11) | 90% | PS, HCM, ASD, VSD |
| Holt-Oram (TXB5) | 90% | ASD, AVB, AVD | |
| Alagille (JAG1) | 95% | PBS, FT, IAC,VSD | |
| SDM of Marfan (FBN1) | Frequent | AR, Ao Aneurysm, MPro |
Abbreviations:
- Ao Aorta
- AoC aortic coarctation
- AR aortic regurgitation
- AoS aortic stenosis
- AVB atrio-ventricular block
- ASD atrial septal defect
- AVD atrioventricular defect
- BAo bicuspid aorta
- FT Fallot’s Tetralogy
- HCM hypertrophic cardiomyopathy
- MPro mitral prolapse
- PBS pulmonary branchstenosis
- PDA patent ductus arteriosus
- PS pulmonary stenosis
- PTS pulmonary trunk stenosis
- RAA right aortic arch
- CTA Common truncus arteriosus
- VSD ventricular septal defect
Murmurs are the noises produced by turbulent blood flow in the heart and vessels and/or vibrations of cardiac structures. To define them well we must refer to all of their characteristics:
- The phase of the cardiac cycle in which they occur: systolic, diastolic, systodiastolic or, continuous. Systolic murmurs can be organic or functional.
- The duration within the phase of the cardiac cycle in which the murmur occurs. This can be: precocious (proto-), medium (meso-), late (tele-), or all-encompassing (holo- or pansystolic.)
- 3. The morphology or form in which a murmur goes from the minimum intensity to the maximum or vice versa. This could be:
- Elective, in crescendo-decrescendo, rhomboidal or in diamond. When the breath is increasing until it reaches its maximum intensity and then gradually decreases until it disappears. It is typical of sigmoid valve stenosis – audible in the apex.
- Regurgitative or pansystolic: constant or rectangular intensity, typical of ventricular septal defect and regurgitation of atrioventricular valves.
- Its intensity, which is measured on a scale of 1 to 6, where 1 is the least audible, above 4 is palpable, also known as ‘a thrill’, while the most intense can be auscultated without the need of the stethoscope.
We use the term organic murmur for a pathological murmur derived from an intracardiac or vessel anomaly, which causes a turbulent flow. It is necessary to differentiate them from the typical innocent murmurs of a healthy child.
INNOCENT MURMURS
The denomination of INNOCENT MURMUR (Evans, 1947) is preferred because it clearly notes the absence of pathology. Their cause is not known with certainty, although they are influenced by higher heart rates in the child, greater elasticity of the myocardium and smaller distances between the heart and thorax, which can all cause the sound of blood flow to be heard.
Given that they are the murmurs that the paediatrician will hear most often, it is essential that he or she knows them and can identify them and differentiate them from pathological murmurs.
Their characteristics are:
- They are short, never holosystolic. R1 will be clearly identified.
- Their intensity is between grades 1 and 3, therefore, they do not present thrill.
- Most of them, like organic murmurs, increase in intensity with the presence of fever, anaemia or high cardiac output.
- They have a defined location with little irradiation.
- They change intensity when changing the position of the patient.
Still’s(vibratory) murmur is the most common functional murmur in children (occurring in 75-85% of school-age children). It can be auscultated at any age, from early infancy through to adolescence, but it is more frequently found between the ages of 2 and 6. It is a murmur of medium-low tone, of moderate intensity (grade 2 or 3 out of 6), confined to the first half of the systole and with maximum auscultation in the left sternal border and apex. It is best heard in the decubitus posture and often decreases in intensity and changes tone with postural changes. The most characteristic feature of Still’s murmur is its musical or vibratory tone.
Other innocent murmurs are: the venous murmur (present at the jugular junction, in the innominate or subclavian arteries with the vena cava, or in the first intercostal space, which disappears in the supine position), the murmur from stenosis of pulmonary branches in the new-born (more audible in the armpits, which disappears during the first months of life), and the carotid murmur from the junction of the supra-aortic vessels with the aortic arch (aortic stenosis must be ruled out).
The diagnosis of innocent murmurs in asymptomatic children must be clearly differenciated frompathological murmurs due to ventricular septal defect (VSD),subaortic stenosis and hypertrophic cardiomyopathy, although these do tend to be rougher and havethrill.
If you want to know more about heart murmurs:
In the neonatal stage, the symptoms and clinical signs of congenital heart disease (CHD) can vary greatly depending on the type of malformation. One must be bear in mind that some new-borns (NB) may not have a heart murmur at birth or show clinical signs of complex cardiac malformations because the pressures between both ventricles are of equal pressure at birth. As the pulmonary resistances decrease and/or the structures of the foetal circulation are closed, the CHDs become evident. At present, with the prenatal diagnosis of heart disease, the management of these patients is being changed. Now, after diagnosis, the NB is delivered in hospitals that have pediatric cardiology departmentsas well as neonatal and paediatric cardiac surgery.
The presentation of neonates with some type of heart disease without prenatal diagnosis, are classified into two clinical forms; the first as shock and the second in the form of cyanosis. Nevertheless, some neonates have mixed forms, and the analysis of the combination of clinical presentations makes it possible to suspect the type of heart disease.
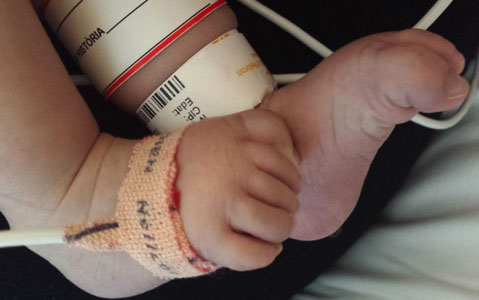
In general, almost all serious heart diseases have symptoms coinciding with the closure of the ductus, either because it maintains systemic output (the closure causes shock because the descending aorta is left without flow – these are ductus-dependent heart diseases of aortic or systemic output) or it maintains pulmonary output (the closure causes intense cyanosis, so these are ductus-dependent heart diseases of pulmonary output).
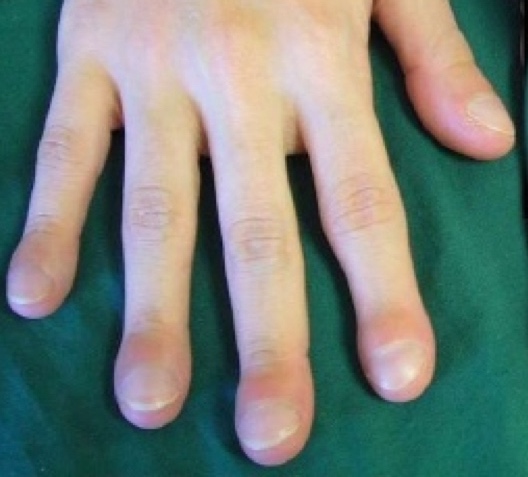
Image of acropachy in fingertips.
As years pass, a sustained left-to-right shunt causes an increase in pulmonary pressure and histological changes of the pulmonary vessel thereby generating pulmonary hypertension. Over time, this increase in flow causes an increase in the thickness of the tunica media (muscular middle layer) and, consequently, an increase in pulmonary resistance which becomes irreversible. This is known as Einsenmenger’s Syndrome, where the shunt remains irreversibly inverted, causing cyanosis (right-to-left shunt). Another secondary complicationof the shunt is the risk of endocarditis. In CHDs that cause cyanosis, there is a compensatory increase in the production of haemoglobin, causing polyglobulia, which, in turn, causes problematic abscesses due to hyperviscosity of the blood. The bony deformations of the fingers (acropachy or “drumstickfingers”) are deformations of the distal phalanges of the fingers and toes caused by hyperproduction of blood in the bones in those locations.
Presently, interventional catheterization is solving simple and isolated heart problems and resolving the complications that appear in the follow-up, thereby resulting in a decrease in the number of surgical procedures.
There are specific devices for closure of septal defects, both interauricular and interventricular, patent ductus arteriosus or fistulas. Openings of narrow vessels can also be performed as in aortic coarctation or valvuloplasties for valve stenosis (including foetal intervention) and, more recently, interposition of stents by catheterization for the opening of vessels and valves in pulmonary and aortic positions.
The first surgical procedure for a congenital heart disease was performed in 1938 in Boston by Dr. Robert Gross and consisted of the closure of a ductus arteriosusby lateral thoracotomy. Link.
In 1958, the appearance of the membrane oxygenator brought about a radical change in surgery. It allowed the surgeon to “stop” the heart and keep the body oxygenated with extracorporeal circulation (ECC) while the procedure was being performed. Link
Progressive technological development allowed the appearance of neonatal cardiac surgery in the 1980s. In 1975, Dr. Adib Jatene successfully performed an anatomical correction on a one-year-old child with a transposition of the large arteries (an inversion in the position of the vessels, so that the aorta is connected to the right ventricle and the pulmonary artery to the left). Dr. Aldo R. Castañeda began to use this technique in neonates from the late 1980s.
Of all the techniques, the most recently developed and the one which carries most risk, has been the surgery performed on children affected by Hypoplastic Left Heart Syndrome (HLHS) known as Norwood surgery. For new-borns that are born with severe underdevelopment of the left ventricle or of the aorta, the surgery consists of joining the mini aorta and the pulmonary artery to form a single vessel, disconnecting the pulmonary branches and maintaining the flow through a tube between the pulmonary branches and the right ventricle (or with the aorta depending on the technique). The development of prenatal diagnosis has reduced the incidences of HLHS and the cases that are operated on have a chance of survival that varies between 60-80%, depending on the hospital.
Cardiac surgeries for congenital heart diseases are divided into closed surgeries (without the need for extracorporeal circulation, usually by lateral thoracotomy) and open (with ECC). They are usually named after the surgeon who performed the technique for the first time. If the patient has two well-functioning ventricles of similar size, and a blood vessel of adequate size, a corrective or biventricular surgery is performed (trying to make the heart as similar as possible to a normal heart), where the good vessel will remain as an aorta and a prosthesis (tube) is placed in the pulmonary position. The tubes used can be homografts (aorta or pulmonary arteries from corpses,which are processed in a tissue bank, denatured and cryopreserved), biological (xenografts, of animal origin or through genetic engineering) or made from synthetic material. All these tubes have the disadvantage of short durability, the need to be small in size (adequate to the weight of a small child) and also that they must be changed as the child grows. When there is no possibility of performing biventricular surgery due to hypodevelopment, absence of a ventricle, or atresia of one of the atrio-ventricular valves, univentricular surgery (also called Fontan surgery) is performed. Here the venae cavae are connected by a tube (usually made from a Goretex® prosthetic material or ePTFE) to the pulmonary arteries, leaving only a single ventricle as a heart.
It is essential that the pulmonary branches have an adequate level of development for the general success of the surgery. If there is severe hypoplasia of the pulmonary branches a complete surgical correction can not be offered.
Another way of naming surgeries is to classify them as anatomical surgeries (correction of a defect according to its anatomical problem) or as physiological surgeries (surgical corrections that let the heart function correctly, although its anatomical defecthas not been corrected).

These heart diseases are the most frequent and can be defects at the atrial, ventricular or vascular level.
Included are: defects at the interatrial level (ASD), atrio-ventricular defects (known as the AV channel), septal defects at the interventricular level (membranous VSD if it is from the upper part of the interventricular septum or muscular VSD if it is from the lower part) and patent ductus arteriosus.
They cause an increase in pulmonary flow that triggers heart failure. Increased flow to the pulmonary circuit causes an imbalance between the pulmonary and systemic flows (Qp/Qs), and so more blood goes to the lungs.
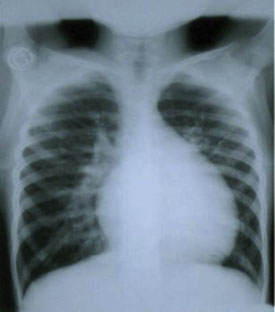
The symptoms are heart murmurs and signs of heart failure while a chest X-ray shows increased pulmonary flow.
The diagnosis is made by echocardiography, and rarely requirescardiac catheterization (only for closure of defects, called interventional catheterization). Sometimes other imaging techniques such as CT Angiography or MR Angiography are performed, especially to see the anatomy of the blood vessels that leave the heart (pulmonary arteries or supra-aortic trunks). Moreover, in every child with suspected cardiac lesions, an ECG must be performed to confirm the heart’s rhythm, whilst also informing about the overload of the cardiac cavities and any repercussions that may stem from it.
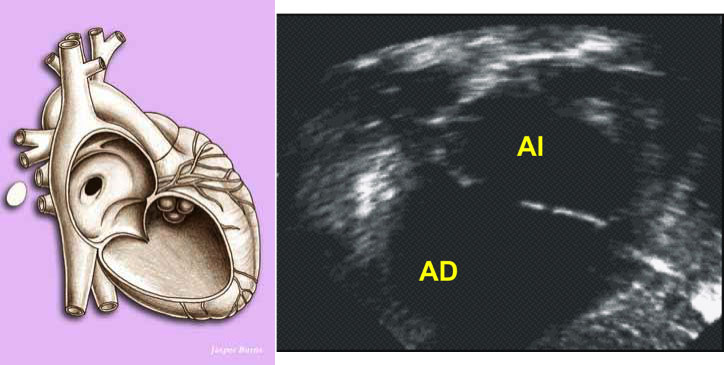
The atrial septal defect (ASD) is a defect of the interatrial septum. The most frequent is the one located centrally, Ostium Secundum ASD. In small children, it must be distinguished from a permeable foramen ovale. The defect produces a passage of blood from the left atrium to the right, causing an overload in the right-hand cavities, and therefore more blood going into the pulmonary circuit. As there is low pressure in the area of the atria, a murmur is produced by the blood that passes through the pulmonary artery, causing a relative pulmonary stenosis. It is treated through closure by cardiac catheterization with various devices. At present, if the defect is very wide, the child is very small, or the edges of the atrial tissue that surround the defect are small,surgery is performed (which involves placing a patch and is performed with extracorporeal circulation).
There are other ASDs in other locations that can not be closed by catheterization, which necessitate surgical closure: those located in the upper part of the septum called sinus venosus ASD, which is sometimes accompanied by abnormalities of the pulmonary veins draining into the right atrium, and Ostium Primum ASD (a defect of the endocardial cushions), located in the lower part of the septum and that is usually associated with abnormalities of the mitral valve (holes in the mitral leaflets which are called “clefts”).
The most common ventricular septal defect (VSD) is located in the lower part of the interventricular septum. It is known as muscular VSD and it usually closes spontaneously. If it is very extensive and requires closure, it can be done through catheterization by the placement of a device or, more rarely, by cardiac surgery (with the placement of a patch). The VSDs located in the upper part, below the arterial vessels, require surgical closure more often. At the present moment, they cannot be closed by catheterization, since they are very close to the aortic valve and can damage the conduction of the heart beat, given that the the bundle of His (which conducts the impulses of the heart beat to the ventricles) is found in the subaortic zone andwhen damaged produces atrioventricular blockage. VSDs cause overloading of the left-hand cavities, and if they are not closed by an attempted medical treatment, surgery is performed. The increased flow at the lung level causes pulmonary hypertension (PHT) sooner than with ASD and must be operated on within the year (maximum 2 years). Sometimes there are multiple VSDs, or are those too large and inaccessible for the surgeon. In these cases, a pulmonary cerclage (a band) can be placed as palliative surgery in the hope of closing some or all of the VSDs and then proceeding to remove the cerclage and close any remaining ones.
Patent ductus arteriosus also causes overload of the left ventricle, particularly if it is of a large size and the amount of blood which runs through it is high. Currently the closure is done by catheterization using devices, even in the prenatal period. It is necessary to differentiate the ductus of the premature new-born from the normal new-born, in these cases medical treatment is performed with paracetamol or ibuprofen before the surgical closure.
Down syndrome is specifically associated with an atrio-ventricular defect, from its mildest form as the defect of the lower interatrial septum (Ostium Primum ASD) to its most complex form, as complete atrio-ventricular septal defect with a common atrio-ventricular valve. It requires corrective surgery at about 6 months old, it is characteristic to see a left axis alteration in the ECG (because it is a defect located in the centre of the heart, where the bundle of His is located; the area of ventricular conduction). In addition, Down Syndrome is usually associated with ductus arteriosus.

Complete atrio-ventricular defect image
These are obstructive lesions that cause obstruction to the flow of blood at the entrance or exit to one of the ventricles. These can include:
Pulmonary stenosis (if it is very severe and associated with a septal defect it can cause cyanosis due to right-to-left shunt)
It can be subvalvular, valvular or supravalvular (very often associated with syndromes such as William’s Syndrome). Valvular pulmonary stenoses are usually solved by catheterization with a pulmonary valvuloplasty. Usually subvalvular and supravalvular stenosis require corrective surgery. There are severe forms of hypodevelopment of the right ventricle and the tricuspid valve, called hypoplastic right ventricle syndrome (or pulmonary atresia with integral septum). The management in these cases is different from a single lesion of the right ventricle outflow tract.
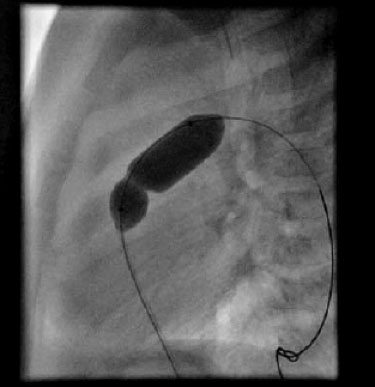
Image of a pulmonary valvuloplasty
Aortic stenosis (according to the degree of obstruction, they are labelled as light, moderate or severe)
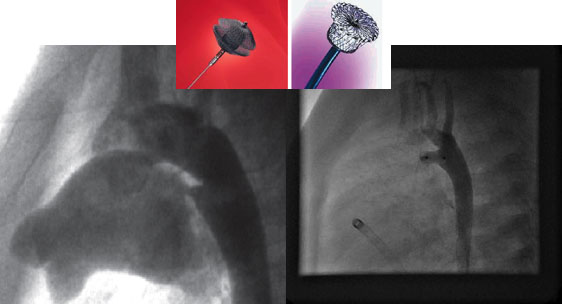
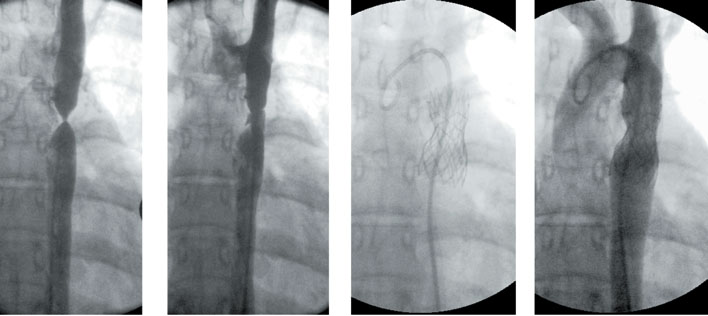
Depending on the location of the narrowing area, they can be subvalvular (if they are localized, they are usually due to an impeller. It can that be the entire outflow tract of the left ventricle, encompassing the aortic valve ring, which complicates its treatment); valvular or supravalvular (characteristic association with the William’sSyndrome, which affects the entrance of the coronary arteries). Their treatment will be determined by the degree of narrowness and its location. In valvular forms the first thing is to perform an aortic valvuloplasty (using balloon dilation by catheterization, as seen in the next diagram) and wait for the child to grow, eventually placing a mechanical prosthesis when feasible (ideally after growth is complete, from around 12 years old).
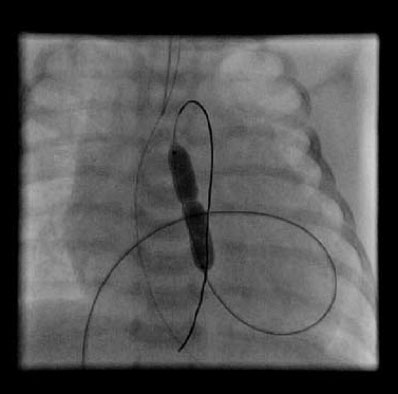
Image of an aortic valvuloplasty
Secondly, a surgery is performed which is known as the Ross technique, or pulmonary autograft in the aortic position (the patient’s native pulmonary valve is placed in the aortic position, a tube is placed in the pulmonary artery that will require replacement as the child grows, but pulmonary stenosis is always better tolerated than aortic stenosis)
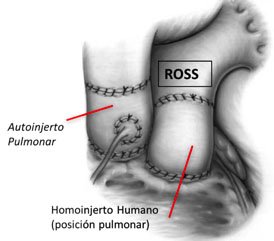
Image of Ross surgery
There are forms of hypodevelopment of the left ventricle with lesions at various levels, the association of aortic coarctation with bicuspid aortic valve, aortic stenosis and mitral valve with a single papillary muscle (parachute valve) is known as Shone Syndrome and develops the child requires interventions at other levels besides on the aortic valve. The most serious form of lesions on the left side of the heart ishypoplastic left heart syndrome (HLHS), which is included in the forms of univentricular heart disease.
Aortic coarctation (narrowing of the exit of the subclavian artery, in the descending aorta, causes arterial hypertension above the obstruction and a decrease in pulsein the area below the narrowing)
In new-borns and infants, it is treated first with surgery and resection in the area of narrow tissue, whereas older children and adults can be treated by catheterization (angioplasty dilatation with the placement of a stent in the narrow area).
Others: Mitral stenosis is rarely found in an isolated form in children, is usually associated with other abnormalities of the left side of the heart. Other abnormalities of the left heart, such as pulmonary vein stenosis, are also uncommon.
Cyanosis is the bluish discoloration of the skin and occurs when the concentration of reduced haemoglobin (Hb) in the blood is greater than 5g/dL. It is necessary to differentiate cyanosis from hypoxia. Hypoxia is a decrease in the concentration of oxygen in the blood. Not every hypoxic child is cyanotic or vice versa. A new-born may be hypoxic and not cyanotic in cases of severe anaemia, since the amount of reduced Hb may be low. Conversely, it may be cyanotic without hypoxia, as would be the case with alterations of haemoglobin, such as in congenital hemoglobinopathies, where the patient will be blue but the oxygen content is normal.
In most cases, congenital heart disease patients with an oxygen saturation (SaO2)
of greater than 65-75% are not hypoxic, so their breathing is normal (no polypnea or respiratory difficulty) unlike patients with hypoxia due to respiratory disorders. If cyanosis appears abruptly (after closing the ductus) it can cause polypnea and shock due to tissue hypoxia (and elevation in levels of lactic acid).
To checkif the cause of cyanosis in the new-born is due to congenital heart disease, a hyperoxia test is performed, where 100% oxygen is administered and the blood is checked to see if SaO2 increases by more than 10% or if the partial pressure of oxygen(PaO2) increases by more than 20-30 mmHg. If this is the case, it is concomitant respiratory disease. If there are no changes in SaO2 or PaO2, we have cyanotic heart disease, although occasionally the new-born presents a situation of persistent foetal circulation (PFC) which is persistence of elevated pulmonary pressure, typical of the foetus, which involvesright-to-left shunting through a remnant of the foramen ovale or a ductus arteriosus. In these cases, the obstetric history helps towards making a diagnosis.
When noting a difference in SaO2 between the upper-right extremity and the lower extremities PFC (persistent right-to-left shunting through the ductus arteriosus) should be suspected.
Recommended readings:
Cyanotic congenital heart disease can be caused by 2 mechanisms:
- Due to obstruction to the right heart associated with a defect that allows a right-to-left shunt through it, such as: pulmonary atresia with VSD, pulmonary atresia withintact ventricular septum (with right-to-leftshunt through the oval hole), tricuspid atresia, or single ventricle with pulmonary stenosis. In these cases, the pulmonary flow is decreased because there is a percentage of blood that does not pass into the pulmonary circuit, but rather goes directly to the aorta (systemic circulation) (having a pulmonary-systemic flow ratio[Qp/Qs] of less than 1).
- Due toparallel circulation as in the d-transposition of large arteries, where there is a change in the arrangement of the blood vessels, causing the blood that goes to the pulmonary circuit not to go to the systemic circuit, therefore requiring a communication so that the oxygenated blood reaches the aorta. Usually after closure of theductus arteriosus, intense cyanosis appears. The amount of blood that goes to the pulmonary and systemic circuits is almost equal, but in parallel.
Mixing is another important concept in CHD. There may be CHD with a right-to-left shunt, but without cyanosis because the mix at the intracardiac level is high and the amount of blood that reaches the pulmonary circuit is adequate (eg. a single ventricle without obstruction to pulmonary flow or an atrio-ventricular defect) or, when the degree of narrowing of the right ventricle outlet tract allows sufficient blood to pass to the pulmonary circuit, where we describe the patient as being “balanced” (without hypoxia or severe cyanosis).
In cases of severe pulmonary obstruction without right-to-left shunt, the heart will try to compensate for the obstruction and if it fails to do so, it will cause shock, as the blood does not reach the systemic circuit.
Image taken from http://www.chd-diagrams.com/heart-disease/114
Of all the heart diseases that present cyanosis, tetralogy of Fallot(TOF) is the most common. It represents 5-8% of all congenital heart diseases. There is a broad spectrum of types, and this section will focus mainly on the classic form, finishing with a look at some special characteristics of pulmonary atresia with VSD and TOF with absence of the pulmonary valve.
22q11 microdeletion and trisomy 21 are associated with TOF. Between 15 and 25% of patients with TOF present 22q11 microdeletion, also known as Catch-22 syndrome (cardiac defects, facial anomalies, thymic hypoplasia, cleft palate and hypocalcemia).
The four basic anatomical elements are:
- Anterior and cranial deviation of the infundibular septum, which causes stenosis at this level, accompanied to a greater or lesser degree by hypoplasia of the pulmonary valve, trunk and branches. What indicates the greatest severity and worst prognosis in this heart disease is the degree of hypoplasia at this level.
- Wide subaortic ventricular septal defect (VSD).
- Overriding aorta positioned directly over a VSD which links both ventricles.
- Right ventricular hypertrophy, caused byright ventricular outflow tract obstruction (RVOTO)
PHYSIOLOGY
The ventricular septal defect is wide, so the direction and magnitude of the shunt through it depends on the degree of pulmonary stenosis, which also tends to be progressive. Often the stenosis starts off mild, with a predominance of the left-to-right shunt, sometimes with symptoms of heart failure, known as pink tetralogy of Fallot. As the disease progresses and the stenosis becomes more severe, the shunt direction is reversed and cyanosis appears.
The clinical features vary according to the degree of pulmonary stenosis, with 25% showing cyanosis at birth. In the older child with uncorrected TOF and severe cyanosis, the “squat” posture, that favours left-to-right shunting by interventricular communication, increases the pulmonary flow and improves hypoxemia.
Hypoxemic crises are sudden episodes of cyanosis, triggered by crying, feeding or defecation, accompanied by polypnea and loss of consciousness. Hypoxemic crises may also appear during certain situations such as fever or intense exercise and are due to a critical decrease in the flow of blood through the pulmonary infundibulum, presenting a characteristic disappearance of the ejection murmur. They can appear in children with or without previous cyanosis. They are a clinical sign of severity and require urgent surgery.
The examination will showprevailing cyanosis, although initially it may be absent or very light. In cardiac auscultation, there is a systolic murmur in the upper-left sternal border, which is more holosystolicwhen the stenosisis less severe. Typically, this murmur diminishes or disappears during hypoxemic crises. The pulmonary component of the second noise is absent or very weak.
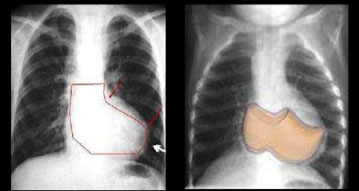
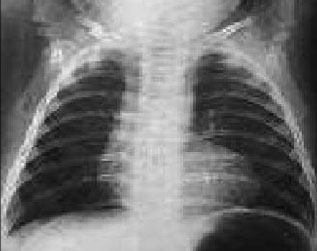
In the chest X-ray, the cardiac silhouette is normal or small with a raised cardiac apex and a reduced pulmonary arterial cone (resulting in a clog-shaped heart, see next image). The pulmonary vascularization is either normal or diminished.
Echocardiography is the diagnostic method of choice and in most cases it provides the necessary information for surgical correction. It allows the visualization of the subaortic VSD, the characteristics of the shunt through it, the percentage of aortic overriding, the location and amountof pulmonary stenosis, the size of the pulmonary ring, trunk and main branches as well as the existence of any associated lesions.
TREATMENT
In exceptional cases, they may require treatment in the neonatal period, in new-borns with severe pulmonary stenosis that present cyanosis with hypoxemia when the ductus is closed. In these cases, the use of prostaglandins and early surgery is obligatory. The majority of patients do not require medical treatment pending surgery, although beta-blockers are sometimes used to relax the pulmonary infundibulum and prevent hypoxemic crises.
Hypoxemic crises are a medical emergency, whose treatment consists of oxygen therapy, the genu-pectoral position, rapid fluid administration, morphine chloride, and, in very severe cases, vasopressor agents (ideally phenylephrine which is a pure alpha-adrenergic agonist, without beta-adrenergic effect, which may worsen the crisis) or intravenous esmolol (an ultra-short-acting beta-blocker). Its prevention consists of avoiding the irritability of the child, including the administration of sedatives, and avoiding constipation. Severe and/or progressive cyanosis and the presence of hypoxemic crises are an indication that urgent surgical treatment is necessary.
The usual age for elective correction is between 3 and 6 months. It consists of closing the ventricular septal defect with a patch and correcting pulmonary stenosis with various techniques. The most frequently used is the opening of the infundibulum and the pulmonary ring by using a patch that extends to the origin of a pulmonary branch (a transannular patch). The price that must be paid with this technique is the progressive appearance of pulmonary insufficiency with inevitable dilation and dysfunction of the right ventricle in the future. The current tendency is to preserve the function of the pulmonary valve as much as possible.
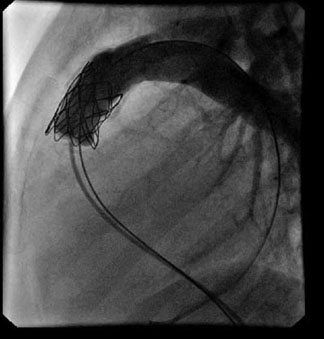
Occasionally, due to age or unfavourable anatomy, complete correction can not be performed, and palliative procedures such as a modified Blalock-Taussig (BT) shunt (surgical interposition of a 4-5mm tube between the aorta or subclavian artery with the pulmonary artery) or the placement of a stent in the ductus of the new-born through catheterization.
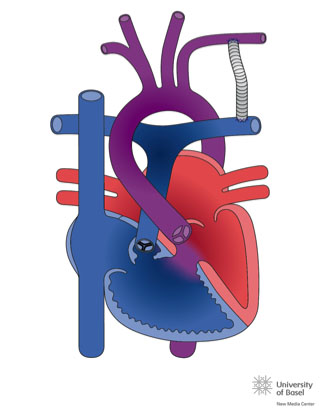
Image of modified left Blalock-Taussig shunt. Taken from http://www.chd-diagrams.com/heart-operation/155
The long-term prognosis of classic TOF is favourable, the overall rate of survival to 30 years old is 90%, although the need for new surgical interventions or interventional procedures in the pulmonary branches is frequent. In the long term, the main problem is pulmonary insufficiency, a consequence of the transannular patch, which can be treated by implanting a prosthesis in the pulmonary position through surgery or, more recently, percutaneously.
Pulmonary Atresia with VSD is considered the most severe form of the Fallot spectrum, although embryologically it may have another origin. From an anatomical point of view, the intracardiac anatomy is similar to that of a large subaortic VSD, aortic overriding, and right ventricular outflow tract obstruction. The diagnostic and therapeutic complexity of this entity is indicated by pulmonary circulation.
The best case scenario is where the pulmonary branches are normally formed and are supplied by a ductus. At the worst end of the spectrum, the central pulmonary branches do not exist or are extremely hypoplasic and the pulmonary circulation comes from systemic-pulmonary collateral branches, coronary branches, or from bronchial circulation.
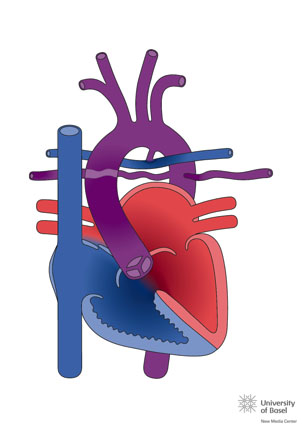
With the morefavourable forms, treatment consists of stabilizing pulmonary flow in the neonatal period using a BT shunt or by implanting a stent in the ductus. in a second surgery, the definitive treatment is in the closure of the VSD and the interposition of a valved conduit between the RV and the pulmonary arteries (known as the Rastelli technique). These patients must be operated on several times to replace the conduits. The average lifespan of the conduits during childhood varies according to the type used, but they usually last an average of around 4 years.
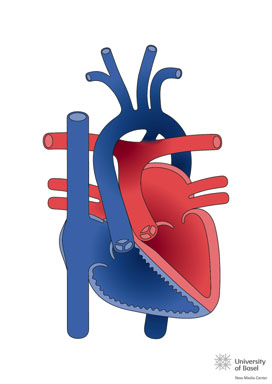
Transposition of the great arteries (TGA), also described as complete transposition of the great arteries, is a congenital heart malformation characterized by atrio-ventricular concordance and ventriculo-arterial discordance. Inthe TGA the morphologically right atrium connects with the morphologically right ventricle, from which the aorta originates (entirely or predominantly), while the morphologically left atrium connects with the morphologically left ventricle, from which the pulmonary trunk originates (entirely or predominantly).
The predominant physiological anomaly in the TGA is that the systemic venous return recirculates to the systemic territory through the right ventricle (RV) and the aorta, while the pulmonary venous return recirculates back to the lungs through the left ventricle (LV) and the pulmonaryartery. Therefore, whereas in the normal heart the systemic and pulmonary circulations are connected in series, in the TGA they function as two separate circuits, in parallel.
The presence of cyanosis on the first day of life suggests a diagnosis of TGA, and if the interventricular septum is intact, the cyanosis can already be severe in the first hours of life. When there is obstruction in the pulmonary outflow tract, the cyanosis is more prominent, while it canbe less apparent or even absent in patients with a large VSD (in these cases the signs of congestive heart failure are prevalent). A new-born with TGA is usually visibly cyanotic and has a discrete tachypnea, without frank breathing difficulty. Cyanosis does not vary with crying or oxygen administration. Usually no murmurs can be heard unless there is obstruction in the outflow tracts. After the first two weeks of life, in untreated patients, there is usually a failure to thrive. This is related to the decrease in food intake combined with the increase in caloric requirements due to the higher amount of respiratory work.
Chest X-ray and ECG may help in the diagnosis phase, but their findings are nonspecific. The ECG usually shows the normal right predominance while the X-rayshows a slight cardiomegaly with the vascular pedicle narrowed (its silhouette having an oval shape) with normal pulmonary circulation.
The echocardiogram is the definitive imaging method in diagnosis, providing accurate information on the anatomical and functional details of the heart disease.
The initial objective in managing the TGA is to ensure an acceptable circulatory mixture. The presence of an atrial or ventricular septal defect that provides a satisfactory mixture with adequate arterial oxygenation allows the planning or postponing of a subsequent corrective surgery without the prior need for palliative procedures.
An intravenous drip of prostaglandin E1 is initially administered to maintain the patent ductus arteriosus and improve oxygenation (dose: 0.01-0.1 µg/kg/min), and maintenance of the acid-base balance, temperature, and correction of hypoglycaemia are all essential. In new-borns that areseverely cyanotic or acidotic after the perfusion of prostaglandin E1,balloon atrial septostomy (or Rashkind’s procedure) should be performed for stabilization. It is currently performed in the neonatal unit, under echocardiographic control (in some hospitals it is still performed in the Low Hemodynamics Unit control by fluoroscopy, but it requires the transfer of a new-born which is usually hemodynamically unstable). It consists of the introduction of a specific catheter which has a balloon that is inflated on the left atrial side which, when stretched, causes a rupture of the septum of the fossaovalis, creating an atrial septal defect.
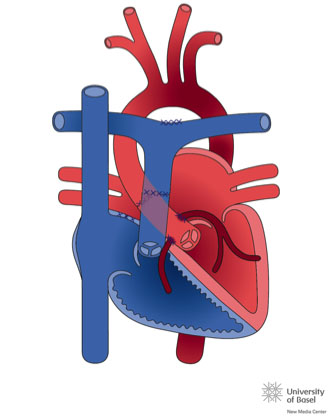
The arterial exchange operation or arterial switch (Jatene technique) is the procedure of choice for the surgical correction of this heart disease at present.
Jatene surgery. Image from http://www.chd-diagrams.com/heart-operation/171
The surgery is preferably done within the first two weeks of life. The operation consists of sectioning the aorta and pulmonary artery just above the aortic sinuses, and re-anastomosing them into their correct anatomical positions, passing the ascending aorta posterior into the position of the pulmonary arteries (known as the Lecompte Manoeuvre). The coronary arteries are removed from their old root together with an aortic wall button and re-implanted in the old pulmonary root. This type of correction has a 90-95% survival rate in uncomplicated TGAs, restoring the normal physiological relationships of systemic and pulmonary arterial flows.
In Switch-type surgical repair techniques (Mustard or Senning procedures), systemic venous return is redirected at an atrial levelto the LV, and pulmonary venous return is directed to the RV. These are rather outdated techniques which are only performed on a few patients with special anatomy.
Senning surgery. Image from http://www.chd-diagrams.com/heart-operation/172/
When there is obstruction at the level of the LV outlet tract, it may be necessary to interpose an extracardiac conduit between the LV and the pulmonary trunk.
All operated patients require a lifetime follow-up. In those who underwent an atrial switch operation, frequent occurrences of arrhythmias and signs of dysfunction of the systemic (morphologically right) ventricle can be present from the second to third decade of life. Patients undergoing an arterial switch operation may present problems related to the appearance of coronary stenosis, dilatation of the aortic root and/or aortic insufficiency.
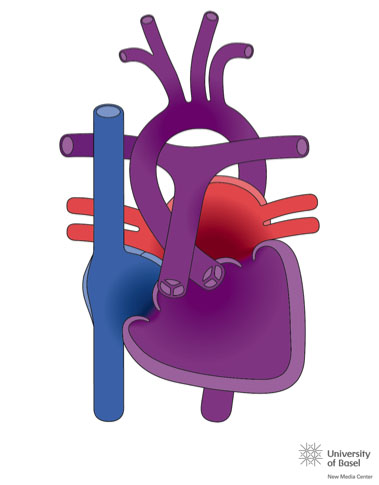
Image from http://www.chd-diagrams.com/heart-disease
The fundamental factor of those complex cardiac anomalies grouped under the term univentricular heart, is that the atrioventricular connection is univentricular, that is, it is completely or predominantly a single ventricular chamber. Most of these hearts have two ventricles, a large one (the dominant ventricle) and a small one which is practically always missing its entrance chamber (the incomplete, hypoplastic or small ventricle). The single ventricle, although only having one functional chamber, has a “bulbo-ventricular” flange,which is the muscular remnant of the other undeveloped ventricle. When there is obstruction to the pulmonary flow, cyanosis appears. If the pulmonary flow is maintained, given the intracardiac mixture, there may be no cyanosis due to the increased pulmonary flow.
In the absence of a ventricle or an atrioventricular valve, biventricular correction can never be established. Either univentricular or Fontan surgery is performedbetween the ages of 4 months and 1 year. This consists of firstly connectingthe superior vena cava to the right pulmonary artery, which is known as the Glenn procedure (http://www.chd-diagrams.com/heart-operation/235)and, subsequently, at around 4 years old, when the lower hemi-body develops, the inferior vena cava is connected to the pulmonary artery.
There are many types of Fontan procedure, which were originally performed by directly connecting the right atrium with the pulmonary artery. Currently the most common technique is the extracardiac total cavopulmonary connection, consisting of the interposition of a Goretex® or PLTE tube between the inferior vena cava and the right pulmonary artery (see image)
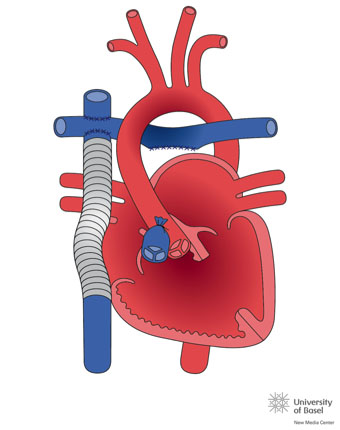
In this surgery there is no right ventricle and so the venous blood reaches the pulmonary circulation, meaning there is only a single circulation. The patient ceases to be cyanotic, although a small amount of the coronary venous blood will drain into the single atrium. The problems in the follow-up stem from the absence of the right ventricle and its pulsatility. There may be an appearance of retrograde hepatic stasis and consequently hepatic cirrhosis ora lesion of the mesenteric flow with the appearance of an intestinal malabsorption syndrome, called protein-losing enteropathy. There may also be deterioration of the single ventricle, sometimes morphologically right, which after causing heart failure would require a heart transplant.
Pulmonary hypertension (PHT) is defined as an increase in average pulmonary arterial pressure (PAP) which is greater than 25mmHg. It is measured by cardiac catheterization, and can be produced by various etiopathogenic mechanisms, as indicated by the classification of pulmonary hypertension (Dana Point 2008; Nice 2013, last update Nice 2018):
- Pulmonary arterial hypertension (PAH) caused by progressive obstruction of the pulmonary arterial bed (including phenomena of cell proliferation and inflammation and thrombosis in the pulmonary arterioles)
- Venocapillary PHT (pressure elevation in the left atrium) due toleft-sided heart disease.
- PHT following hypoxemia and/or lung disease.
- PHT due to thromboembolic obstruction of the chronic pulmonary arterial bed.
- PHT due to diseases of the pulmonary vasculature.
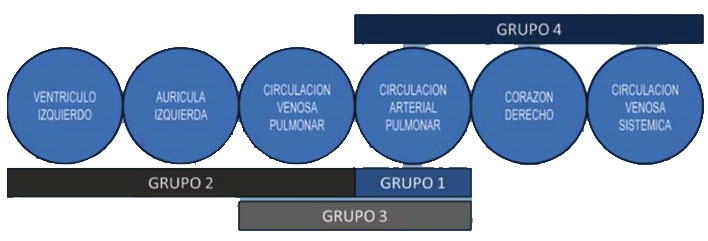
Modified from Saavedra LE et al. Advances Cardiol 2009; 29 (2): 165-178
Congential Heart Diseases will result in PHT mainly through causes 1 and 2. Currently there are multiple pharmacological treatments aimed at avoiding pulmonary vasoconstriction and the remodelling of the pulmonary vessel, and the ways these work are diverse. There are drugs that act on prostacyclines, orendothelin receptor antagonists which cause an increase in cGMP (inhibitingphostodiesterase type 5), but all of them have the effect of dilating the pulmonary arteries. The PHT, if not controlled, evolves and causes hypertrophy of the pulmonary vesselleading to right ventricular failure followed by death. When the progression is very severe and the medical treatment is not stabilized, the patient must undergo a lung transplant. Link
There are an increasingly smallnumber of patients who can not be operated on at the pediatric age, or else, theirsymptomsgo unnoticed and they develop elevation of pulmonary pressure in the follow-up that leads to Eisenmenger’s Syndrome. This consists of a hyperplasia of the pulmonary vascular muscle, generating a fixed elevation of the pulmonary resistance. If the underlying heart disease is a septal defect, the left-to-right shunt is reversed, becoming right-to-left, where the patient becomes cyanotic (having a bluish discolouration of the skin due to an increase in the amount of reduced haemoglobin in the blood) and can even become hypoxic (having a lack of oxygen in the tissues). Hypoxia causes an increase in the production of erythrocytes at the medullary level, which gives rise to polycythaemia. As for the levels of haemoglobin, if they are high, there is an increase in the viscosity of the blood that can cause complications such as embolic phenomena and abscesses.
In the Eisenmenger situation, patients cannot be operated on for their underlying heart disease. It is very important to avoid hypotension, anaemia and other environmental factors that can trigger a crisis of pulmonary hypertension and death. The only treatment options are a cardiopulmonary transplant, or a lung transplant involving a correction of the heart disease, if it is simple enough.
Currently, with the new drugs that control pulmonary pressure, the survival rate of Eisenmenger patients is increasing. The survival rate to 15 years old is 77% and 42% live to 25. The most common causes of death are sudden death (30%), heart failure (25%), and hemoptysis (15%).
Infective endocarditis (IE) is an inflammatory process of infectious etiology that affects the endocardium and/or cardiac valves. The endothelial surface is initially injured by turbulent flow in children with congenital heart disease or,in children without heart disease, by direct injury of the central catheters or bacteraemia. Fibrin, platelets and sometimes red blood cells are deposited on the damaged endothelium, forming an aseptic thrombus. The most common pathogens associated with IE in children are the viridansgroup streptococci and staphylococcus aureus. How it is presented depends on the underlying heart disease, the degree of involvement of other organs (embolization) and the causative agent.
Diagnosis is based on clinical history, physical examination, blood cultures and other laboratory tests, and the echocardiogram. IE has a very variable clinical presentation and there are diagnostic criteria (Duke Criteria) that should be used as a clinical guide to help diagnosis.
It is important to know the prophylaxis of IE in children with heart disease, as well as the criteria for the diagnosis of endocarditis.
Recommended reading:
Myocarditis is an inflammatory process of the myocardium, its incidence is unknown as it can occur without symptoms, and cause chronic dilated cardiomyopathy as it progresses. It may present indifferent forms including: asymptomatic cases, a form similar to acute coronary syndrome that exhibits precordial pain, and a severe form with cardiac dysfunction and even cardiogenic shock, which sometimes require ventricular assistance such as ECMO, or even a heart transplant if there is no recovery or therapeutic response.
Clinical suspicion, the elevation of troponins and echocardiography all help in its diagnosis. Increasingly, cardiac MRI plays an important role in the diagnosis, as it can corroborate the diagnostic suspicion by detecting signs of oedema and inflammation, although the most specific test is the endomyocardial biopsy (EMB). Currently, EMB is performed when the patient does not progress adequately, and can help the treatment. If there are remains of avirus in cardiac tissue, some centres treat it with interferon, and if there are signs of inflammation without a virus, it is treated with immunosuppressants (corticoids and/or mycophenolate).
It is unknown why some children develop myocarditis and progress to dilated cardiomyopathy, and can then relapse. There are data about immunological alterations, for which the administration of Gammaglobulins is added to the treatment of these children at the time of diagnosis.
For more information, we recommend reading the UPIIP Protocols: Acute myocarditis in pediatrics, 2016 (Spanish).
Pericarditis is the inflammation of the pericardium, and its causes can be divided into infectious and non-infectious. Between 40 and 86% of cases are considered idiopathic etiology, although it is believed that many of them are secondary to a viral infection. The inflammation of the pericardium is caused by an infiltration of granulocytes and lymphocytes. Later on, there may be local vasodilation, increased capillary permeability, and massive amounts of protein and fluid in the pericardial space (effusion).
The pathogenesis of the signs and symptoms of pericardial effusion is determined by the increase in intrapericardial pressure, and not only depends on the absolute amount of fluid, but also on the speed with which it accumulates as well as the characteristics of the pericardial fluid itself. Rapid accumulation is clinically less tolerated than slow accumulation, which can allow a large accumulation of fluid without producing symptoms.
Changes in the ECG associated with pericardial disease are the consequence of the patient’s own pericardium and the subjacent myocardium that is also inflamed or compressed.
Pericarditis is the most common cause of ST segment elevation in children. Several phases are defined in its progression. In the initial phase, there is generalized elevation of the ST segment. This phase can last for hours or a few days, and is not constant.
miocardio subyacente también inflamado o comprimido.
La pericarditis es la causa más común de elevación del ST en el niño. Se definen varias fases en la evolución. En la inicial hay elevación generalizada del segmento ST. Esta fase puede durar horas o pocos días, y no es constante.
It is followed by flattening of the ST and the T wave. After this,a diffuse T wave inversion occurs, whichcan last for weeks or months, then finally the T wave is normalized. In cases of significant pericardial effusion, the T waves show frequently low voltages.
The diagnosis is made with a chest X-ray, in addition to the clinical suspicion (fever with precordial pain which is exacerbated when lying face-up and sometimes auscultation of a pericardial friction rub). The X-rayshows cardiomegaly and the echocardiogram, which is usually the best diagnostic tool, confirms the existence of an abnormal quantity of fluid in the pericardial cavity.
The treatment depends both on the causal agent and the amount of fluid. The treatment of pericarditis is principally medical. It is based on rest and anti-inflammatories, in addition to any etiological treatment that may be used. In the case of purulent pericarditis, an invasive procedure isoften required to drain the thick effusion it causes. In tuberculous pericarditis,even with antituberculosis treatment, is difficult to drain the effusion by pericardiocentesis. A pericardiectomy should be considered a priority.
Kawasaki disease (KD) is a self-limiting and acute vasculitis of unknown origin, typical ininfancy, which mainly affects the coronary arteries. The disease is much more frequent in Asia; the incidence in Western Europe is less than 10 cases in 100,000 inhabitants per year, while in Japan it is up to 20 times higher. There is a certain seasonal pattern, with an increase in the number of cases in winter and spring. Occasionally there are peaks of incidence similar to epidemic cycles. Most cases occur between 3 months and 5 years of age. The cause of KD is unknown, but the clinical and epidemiological data strongly suggest an infectious origin.
In the acute phase, systemic inflammation is triggered,causing an increase in lymphocytes(especially B-lymphocytes)thereby activating a‘cytokine storm’, which then generatesa vasculitis called Polyarteritis Nodosa (PAN)which predominantly affects small- to medium-sized arteries. In the most affected vessels, the elastic lamina is destroyed, which causes dilatation, leading to the formation of aneurysms and progressive fibrosis that generates stenosis of the affected vessels.
The diagnosis is based on the existence of fever of at least 5 days and at least 4 of the 5 remaining clinical criteria:
- Bilateral conjunctival injection (non-exudative).
- Alterations in theoral or oropharyngeal mucosa: intense redness and labial fissures, oropharyngeal exanthema of varying intensity, strawberry-like tongue.
- Polymorphous exanthema (morbilliform, or similar to rubella or scarlet fever), which mainly affects the trunk and extremities.
- Tough edema in the hands and feet, potentially accompanied by redness on the palms and soles. Periungual peeling in the second or third week.
- Cervical lymphadenopathy, usually isolated, of more than 1.5 cm in diameter.
In the presence of fever and coronary alterations, the diagnosis can be made with fewer than 4 of these principle criteria. Up to 20% of patients do not meet all these principle criteria and are said to have incomplete KD, which especially affects children under one year of age. In these patients the diagnosis can be delayed and, therefore, there is a greater probability of developing coronary complications.
NOTE: KD should be considered as a diagnosis for infants under 6 months old with prolonged fever of unclear origin and laboratory data about systemic inflammation.
Coronary lesions appear between 1 and 4 weeks after the onset of fever in about 20% of untreated patients and are responsible for most of the mortality and morbidity of KD. Coronary dilatations of variable size can be detected, which mainly affect the proximal portion of the main coronary arteries. They can coexist with areas of stenosis and are susceptible to aortic thrombosis, causing myocardial infarction.
Due to its high availability, sensitivity and specificity, the echocardiogram is the technique of choice to detect coronary alterations. It must be done during diagnosis, at 2-4 weeks and periodically thereafter as a follow-up for patients with aneurysms. Other imaging techniques, such as magnetic resonance imaging (MRI) or multi-slice computed tomography (MSCT), may be useful for detecting distal aneurysms or determining the locations of complications such as thrombosis or occlusion. Conventional coronary angiography provides better defined image of the lesions, but should not be performed until inflammatory process is completely resolved.
Treatment of the acute phase is performed with 2g/kg of intravenous immunoglobulin G (IVIG) in a single dose, preferably before the 10th day of illness, thereby producing a rapid decrease in activity, though this mechanism is poorly understood. Acetylsalicylic acid (ASA) is used in association with IVIG to reduce symptoms and stop inflammatory activity. Initially, anti-inflammatory doses are administered at 80-100mg/kg/24h in 4 doses, and when thepatient has been afebrile for 48-72 hours, the dose is reduced to 3-5mg/kg/24h to maintain its action as an antiaggregant of platelets in the blood.
In patients with no evidence of coronary lesions, antiplatelet therapy with ASA is maintained at low doses for 6-8 weeks, until the erythrocyte sedimentation rate (ESR) is normalized and the echocardiogram confirms the absence of coronary anomalies.
In patients with small aneurysms, antiplatelet therapy is continued until echocardiographic resolution is demonstrated. In the presence of large or very large aneurysms, anticoagulantsshould be added. In cases involving young children, low-molecular-weight heparin should be added instead. 50% of aneurysms spontaneously return within a year or two, especially in children under one year of age, particularly when they are small in size. However, there may be vascular wallthickening and increased arterial stiffness which lead to an increasedrisk of cardiovascular complicationsin adult life.
In older children with coronary lesions, recommendations regarding sporting practice depend on their clinical situation and results of myocardial perfusion tests.
Electrocardiography (ECG) is the process of graphically recording the electrical activity that is generated in the heart. It provides data on cardiac function,rhythm and conduction disorders, mid-cavitary hypertrophy, and helps in the diagnosis of congenitalor acquired heart diseases. However, a normal ECG does not rule out cardiac complications.
In the child, it is necessary to take into account how the characteristics differ from that of the adult, especially those derived from changes in weight, height and heart rate (PR and QT intervals vary with age).Also, as a child matures, initially the right ventricle is predominant and until 12-14 years there are data of hypertrophy of the right ventricle that are considered normal. The heart rate decreases with age, while the duration of the P wave, the QRS complex, and the PR interval all increase.
For the correct interpretation of the ECG, it is essential to know the leads, their spatial orientation and their polarity. The ECG is based on the graphic record of the electrical impulse (vector) that generates a wave, which can be positive (+) if they approach the positive pole of the lead, and a negative wave (-) if it moves away from it. The usual surface ECG consists of 12 leads, 6 frontal leads and 6 horizontal leads (or precordial leads).
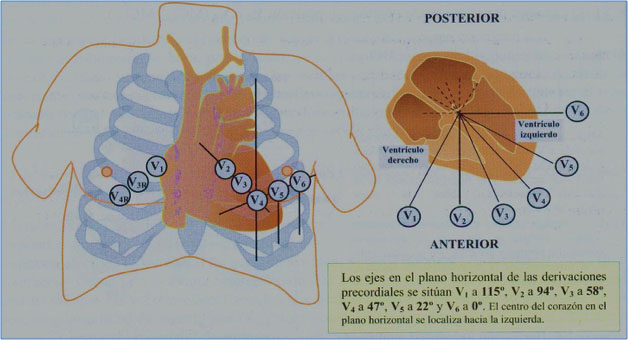
It requires systematic reading after a sequence to obtain a complete study. The following sequence is one of many that can be used:
- Rhythm (sinus or not).
- Heart rate.
- QRS axis.
- Intervals: PR interval, QRS duration, QT interval.
- Segment ST and wave T.
- Study of cavity growths.
Given all the information currently available, we recommend reading the following resources: (Spanish)
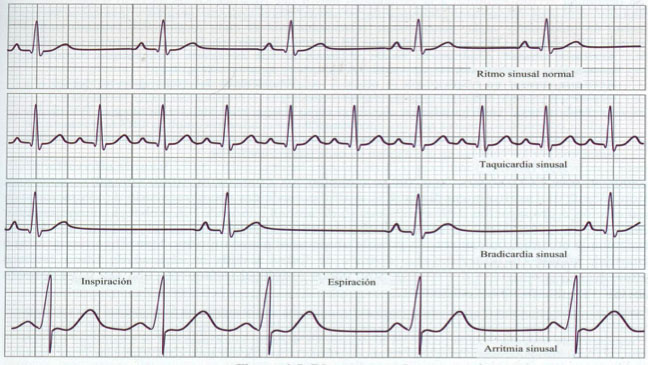
The term arrhythmia encompasses a very broad spectrum of heart rhythm abnormalities. In children, there is usually a physiological respiratory arrhythmia, which can be detected during cardiac auscultation, and is derived from volume changes produced during inspiration and expiration. If this phenomenon is very pronounced, an ECG should be performed to determine if a true arrhythmia is present.
When dealing with an arrhythmia, in addition to the ECG, an adequate anamnesis must be made. Any family history of sudden death or arrhythmia treatment must be known, as well as any symptoms present in the child, such as frequent dizziness, precordial pain, fainting or heart palpitations. The symptoms and signs of arrhythmia are dependent on the effects they exert on cardiac output, the underlying heart disease and the age of the patient.
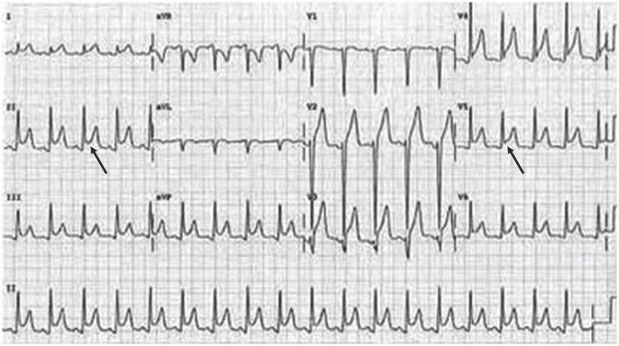
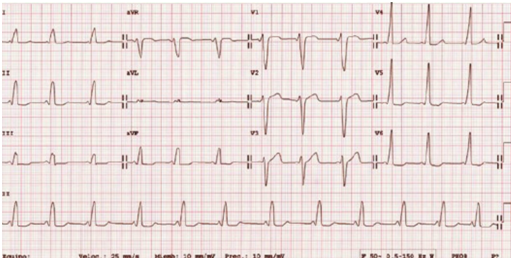
The ECG of a child with Wolf-Parkinson-White syndrome.The short PR interval and the delta wave that causes widening of the QRS (in left precordial, right accessory pathway) can be seen
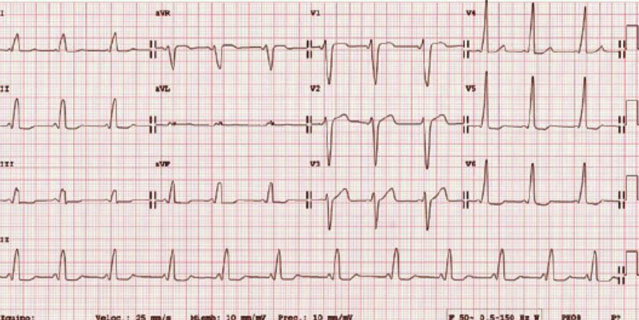
Upon suspicion of arrhythmia, the 12-lead ECG provides information about the patient’s base rhythm, conduction, repolarization, any evidence of hypertrophy, as well as other signs. Alterations in the base rhythm can be very helpful in the diagnostic approach.
Obtaining an ECG during the arrhythmic episode is of great importance for diagnosis. When the arrhythmia is short and sporadic, it is difficult to get an ECG, and so a Holtermonitor may be necessary, which constantly records the rhythm for 24 hours. Currently, there arecardiac event recorders (which record over longer periods, even up to 2-3 weeks) or implantable Holter monitors available for diagnoses, and wearable diagnostic items are also beginning to appear (t-shirts, watches, devices that can be added to a Smartphone, etc).
The most frequent arrhythmias in children are extrasystoles, which are additionalheartbeats that occur outside the normal rhythm. The extrasystoles can be atrial or ventricular, and should rule out the probability of triggering tachycardia, although frequent ventricular extrasystoles may cause concern. When they are of similar morphology (monomorphic) and disappear when exercising they can be considered benign, so in such cases, cardiac pathology must be ruled out and they must be reviewed by a pediatric cardiologist.
Arrhythmias to focus on:
TACHYCARDIA
To manage tachycardia properly,it is necessary to find its origin (atrial, atrioventricular or ventricular node) before starting with the ECG. Atrial tachycardia must be differentiated from sinus tachycardia. ‘Flutter’ or fibrillation are tachycardia seen less frequentlyin children than in adults.
Supraventricular tachycardia (SVT) is produced by two mechanisms:
- Nodal reentry (AV Nodal Reciprocating Tachycardia), seen in older children, by double conduction system within the AV node.
- Reentry by anomalous route (Accessory AV Reciprocating Tachycardia), which is the main mechanism of pre-excitation syndromes (Wolf-Parkinson-White). It is an accessory pathway located around the tricuspid or mitral ring that directly connects the atrial and ventricular muscles. In an ECG one observes an initial widening of the QRS known as the delta wave and ashort PR interval due to the pre-excitation of the ventricle by the accessory pathway. A distinction is made between orthodromic tachycardia (the most frequent, with narrow QRS, no delta wave, and retrograde P) and antidromic (wide QRS, with delta wave, andanterograde P).
- Other causes of tachycardia are due to abnormal automatism (less than 10%), especially in fetal tachycardia. It is produced by impulses in tissues that normally do not have this capacity.
There is a special form of reentrant tachycardia mediated by an accessory pathway with decremental conduction. It is called incessant AV junction reciprocal tachycardia(Coumel type) which is slow (120-180 bpm), persistent, incessant, presents a narrow QRS and is very difficult to control pharmacologically, in most cases requiring radiofrequency ablation treatment.
Supraventricular paroxysmal tachycardia originating in the AV junction are the most frequent forms in adolescents and adults, while tachycardia with reentry by accessory pathway is more often seen in young children. They have a high frequency (200-300 bpm), and present a narrow QRS. They respond to vagal manoeuvres and drugs that slow conduction through the AV node. Clinically, they are well tolerated, but when prolonged, over time they may show signs of congestive heart failure (CHF) (typical in new-borns and small infants). It can also be diagnosed during pregnancy, and the mother treated with drugs that cross the placental barrier.
When QRS is wide, we may be dealing with ventricular tachycardia (VT), which is less frequent than SVT. When facing a ventricular tachycardia with right bundle branch block (RBBB) morphology, it is important to rule out familial catecholaminergic polymorphic tachycardia and arrhythmogenic RV dysplasia. In these cases, the patient should be treated with beta-blockers or sotalol, maybe consider radiofrequency ablation or even the implantation of an automatic defibrillator in cases with risk of sudden death. Cases of VT can be found in patients with long QT syndrome (LQTS), in cardiac postoperative patients with a history of ventriculotomy and in those withmyocarditis or cardiomyopathy.
The treatment’s main objective is to interrupt the arrhythmia, slow down the ventricular response and re-establish an adequate sinus rhythm. Depending on the patient’s clinical situation, Cardio-Pulmonary Resuscitation (CPR) measures may be necessary. If the patientis unstable and with a high hemodynamic compromise, electrical cardioversion should be used first. With SVT, vagal manoeuvres will be performed (the most effective in children is by suffocation with ice), followed by the administration of Adenosine.
ADENOSINE
- Attack dose: 0.1 mg/kg – intra-venous in very fastinfusion.
- If there is no effect, repeat the bolus in increasing doses up to the maximum dose.
In cases of SVT, medical treatment will be maintained (usually with beta-blockers, flecainide and,as a last measure, amiodarone) and, given the low risk of ablation, this procedure is currently performed on patients requiring long-term treatment that have relapses after suppression, or who havefrequent episodes despite the treatment. The ablation consists of applying heat or cold (cryoablation) on the anomalous pathway area to cut the conduction pathway that causes the arrhythmia.
In the case of ventricular tachycardia, the underlying disease must be treated and occasionally an implantable cardioverter-defibrillator (ICD) may be required.
Recommended reading:
BRADYCARDIA
Bradycardia is the low frequency heart rate for a particular age. It is necessary to differentiate abnormal bradycardia from the sinus bradycardia, caused by certain systemic states (hypothyroidism, hypothermia or through drugs). Sinus node dysfunction is very rare in healthy children. It is characterized by sinus bradycardia, chronotropic incompetence to exercise, and sinus pauses. It is typically seen in the postoperative period of corrected congenital heart disease.
Auricular-ventricular block (AVB) is another cause of bradycardia. There are 3 types of AVB:
- 1st degree AVB that does not cause bradycardia, and consists of a lengthening of the PR interval
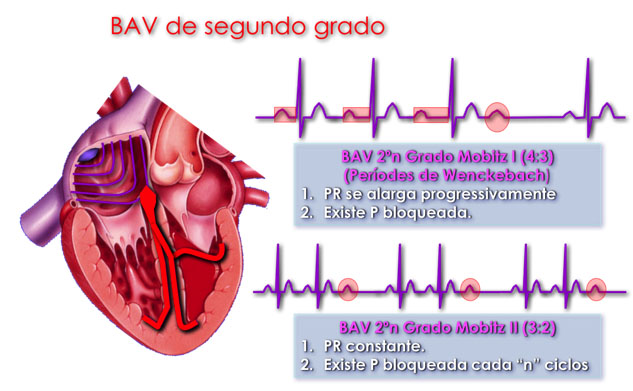
- 2nd degree AVB which is divided into:
-
- type I – which involves a progressive lengthening of the PR interval until a P wave does not conduct (Wenckebach effect).
- type II – where a P wave is constantly followed by a ventricular contraction.
- 3rd degree or complete AVB, which is rare in children (1 in 20,000 live births) but it is potentially lethal. Typically, a dissociation between the atrial and ventricular frequencies is observed. It can be congenital (a child whose mother has systemic lupus erythematosus, or autoimmune disease with anti-SSA/Ro or anti-SSB/La circulating autoantibodies) or acquired (through a complication in cardiac surgery or ablation, or following infection such as viral myocarditis or Lyme’s disease).
Neonatal management of AVB is passedto neonatal admissions for monitoring. When the ventricular rate is low, early use of isoproterenol (0.1 μg/kg/minute) may be useful, but the definitive treatment is the placement of a pacemaker.
Pacemakers are the treatment of choice when facing clinically symptomatic bradycardia. The main objective of the pacemaker is to achieve a heart rate which is as physiological as possible. In small children, epicardial wires are fitted (via thoracotomy) and inolder ones, endovascular cables are implanted (the catheters will need to be replaced as the child grows).
The use of pacemakers and ICDs is becoming increasingly widespread in the paediatric profession, so it is important that pediatriciansknow the precautions patients must follow totake care of their pacemaker (PM). Their activity should only be restricted from contact sports. As for other restrictions, each case depends on an individual’s heart function, rather than on the PM itself.
Prophylaxis of bacterial endocarditis is advisable in patients fitted with endovascular cables. MRI is discouraged, although there are more and more MRI-compatible pacemakers.